








Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkVitae
Print version ISSN 0121-4004
Vitae vol.17 no.3 Medellín Sept./Dec. 2010
INDUSTRIAL FARMACÉUTICA
ACOPLAMIENTO MOLECULAR, 3DQSAR Y DISEÑO DE NOVO DE BENZIMIDAZOLES E IMIDAZOLINAS DERIVADOS DE (S)-ISOTIAZOLIDINONAS COMO INHIBIDORES DE LA PROTEÍNA PTP 1B
MOLECULAR DOCKING, 3D-QSAR AND DE NOVO DESIGN OF BENZIMIDAZOLES AND IMIDAZOLINES (S)-ISOTHIAZOLIDINONES DERIVATIVES AS PTP 1B INHIBITORS
Judith C. GRANADOS R.1; Elsa R. ARIAS P.1; Dency J. PACHECO L.1; Verónica VALDIRIS A.; Ricardo VIVAS-REYES2
1 Grupo de Investigación en Heterocíclicos. Facultad de Ciencias Básicas, Universidad del Atlántico. Barranquilla, Colombia.
2 Grupo de Investigación de Química Cuántica y Teórica. Facultad de Ciencias Exactas y Naturales. Universidad de Cartagena de Indias. Cartagena D. T. y C., Colombia. rvivasr@unicartagena.edu.co.
RESUMEN
Se realizó un estudio tridimensional cuantitativo de relación-estructura (3D-QSAR) con 40 moléculas tipo benzimidazol e imidazolina derivadas de (s)-isotiazolidinonas y su unión con el sitio activo de la proteína tirosina fosfatasa 1B (PTP 1B), utilizando el programa GOLD 3.0. La superposición molecular de los ligandos en la plantilla fue llevada a cabo por el método Database Alignment. El mejor modelo fue el constituido por la combinación de los campos estéricos y electrostáticos de CoMFA, los cuales arrojaron los siguientes parámetros: q2 = 0,659 y r2 = 0,997. Usando el módulo LeapFrog de SYBYL fue posible generar más de 10.000 moléculas nuevas, de las cuales 46 mostraron, teóricamente, un mejor valor de la actividad biológica que su precursora. Los datos obtenidos en el presente estudio podrían impulsar el diseño de nuevos y más potentes inhibidores de la PTP 1B, como agentes para el tratamiento de la diabetes.
Palabras clave: bioinformática, modelos moleculares, diabetes mellitus, benzimidazoles, imidazolinas, inhibidor.
ABSTRACT
A study of the relationship-dimensional quantitative structure (3D-QSAR) with 40 molecules derived from benzimidazole and imidazoline (s)-isotiazolidinonas and their union with the active site of Protein Tyrosine Phosphatase 1B using the program GOLD 3.0 was carried out. The molecular supression of the ligands in the grid was performed by the Database Alignment method. The best model formed by combining the esteric field and electrostatic fields of CoMFA, yielded the following parameters: q2 = 0.659 and r2 = 0.997. Using LeapFrog module of Sybyl was possible to generate more than 10,000 new molecules of which 46 showed theoretically a better value of biological activity than their forerunner. The data generated by this study could promote the design of new and more potent PTP 1B inhibitors as agents for the treatment of diabetes.
Key Words: bioinformatics, molecular models, diabetes mellitus, benzimidazoles, imidazolines.
INTRODUCCIÓN
En las últimas tres décadas, la diabetes mellitus tipo 2 (DM2) se ha comportado como una auténtica epidemia (1). Esta enfermedad aumenta cada año un 6%, y si esta tendencia continúa, en quince años esta porcentaje se duplicará (2). La diabetes tipo 2 afecta aproximadamente al 85% de las personas con diabetes; es un desorden caracterizado por un exceso de glucosa en la sangre y los tejidos del cuerpo y obedece a un defecto en la secreción de insulina, combinado con insulinorresistencia que ocasiona hiperglicemia (2).
Estudios realizados sobre la proteína Tirosina Fosfatasa 1B (PTP 1B) demuestran que un aumento de su actividad en los humanos acarrea insulinorresistencia y diabetes tipo 2 (3). Por otra parte, se ha demostrado experimentalmente que compuestos tipo isotiazolidinona ejercen actividad en cuanto a la inhibición de la proteína tirosina fosfatasa 1B (PTP 1B, crucial en la regulación de la defosforilación del receptor de insulina (4,5) y, por ende, juegan un papel importante en el tratamiento de la DM2. El objetivo de esta investigación es proponer nuevos ligandos capaces de inhibir la PTP 1B, como agentes para el tratamiento de la DM2 por medio de herramientas computacionales.
MATERIALES Y MÉTODOS
Acoplamiento molecular
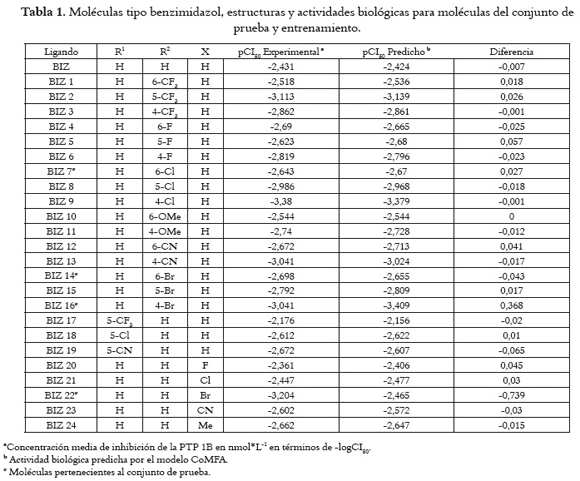
Como base para el estudio se seleccionan los datos comunicados por Douty et al., 2008 (4) y Sparck et al., 2007 (5) sobre la actividad biológica in vitro (CI50 nM) de benzimidazoles e imidazolinas derivados de (s)-isotiazolidinonas, indicados en la tabla 1. Se tomó un conjunto de 40 moléculas para este propósito, se optimizaron usando el método de cálculo HF junto con la base de cálculo 3-21G*, usando el programa Gaussian 03 (6). Se practicó un cálculo de acoplamiento molecular sobre los 40 ligandos optimizados con el modelo proteico de la PTP 1B; la estructura cristalina de esta proteína con código 2VEW se tomó del Protein Data Bank (PDB), utilizando el programa GOLD 3.0 (7), con la función de evaluación GOLDScore, dejando los parámetros del algoritmo genético por defecto.
Conjunto de datos para el análisis
Se tomaron las mejores conformaciones obtenidas del acoplamiento molecular para construir el conjunto de datos, en el que 32 compuestos representaban el conjunto de entrenamiento y 8 pertenecen al conjunto de prueba, estos últimos se eligieron manualmente con el fin de incluir diversidad estructural y un amplio rango de datos de actividad, para evaluar el poder predictivo de los modelos resultantes. Los valores de CI50 (nM) fueron convertidos en sus valores correspondientes de pCI50 (-logCI50) y usados como variable dependiente en el Análisis Comparativo de Campos Moleculares (CoMFA). Los campos de contorno generados a partir del modelo se evaluaron con el fin de relacionar cambios estructurales con modificaciones en la actividad biológica de dichas moléculas.
Alineamiento molecular
Uno de los parámetros ajustables más importantes en 3DQSAR es el alineamiento relativo de todas las moléculas con respecto a una en particular, de tal manera que tengan una conformación comparable y una orientación similar en el espacio (8). El compuesto 15 es el más activo y el más estable farmacológicamente, ya que posee el mejor valor de actividad biológica (22 nM) del grupo de moléculas estudiadas (4). Por esta razón, se utilizó como plantilla para la superposición molecular, asumiendo que posee la conformación más bioactiva de los derivados de (s)-isotiazolidinonas a nivel de sitio activo de la enzima. Cada uno de los análogos se alineó a la plantilla por rotación y traslación para minimizar la desviación cuadrática estándar entre los átomos de la plantilla y los del análogo, usando la opción DATABASE ALIGN en SYBYL (9). Se seleccionó un total de 14 átomos para la superposición. Este fragmento común (en rojo) se aprecia en la figura 1.

Energías de interacción CoMFA
CoMFA muestra los campos estéricos y electrostáticos que rodean al grupo de ligandos y construye un modelo 3D-QSAR correlacionando estos campos 3D con su correspondiente actividad biológica experimental (8).
La energía de campos estéricos (potenciales de Lennard-Jones), y electrostáticos (Potencial Coulombico) se calculó usando un carbono de prueba sp3 y aplicando TRIPOS como campo de fuerza. La energía total fue truncada a ±30 kcal/mol (9).
Análisis de mínimos cuadrados parciales (PLS)
Se aplicó el método de mínimos cuadrados parciales para correlacionar los campos de CoMFA con valores de actividad biológica (10). Para la validación cruzada se empleó el método LOO (por su sigla en inglés Leave One Out), en el que un compuesto es eliminado del conjunto de datos y su actividad es predicha usando el modelo derivado del resto de moléculas del conjunto. Para la validación cruzada se usó un filtro de columna ajustado a 2.0 kcal mol-1. El número óptimo de componentes usado para derivar modelos no validados se definió como el número de componentes que arrojara la mayor validación cruzada y el menor error estándar de predicción (8). Para obtener límites estadísticos confiables en el análisis, se aplicó un método de muestreo (bootstraping) con 100 grupos.
Diseño de Novo
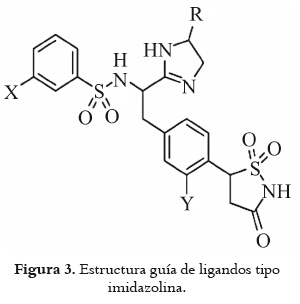
Una vez conocidos tanto el modo de acoplamiento como la relación estructural existente entre los diferentes ligandos y su actividad experimental, se procedió a la optimización de las moléculas con mayor actividad biológica de todo el grupo, a partir de la mejor conformación obtenida en el acoplamiento molecular por medio del módulo LeapFrog de SYBYL (11). El ligando se depositó en una caja cúbica en la que se encontraba la cavidad y fue posible calcular la energía de interacción total entre el sitio de unión y el ligando, al sumar las contribuciones de todos los átomos pertenecientes al ligando. Este módulo es usado para el diseño de fármacos por medio de la implementación de distintas clases de movimientos moleculares. El procedimiento consistió en ejecutar 10000 modificaciones proporcionales a los movimientos JOIN, FUSE, OPTIMIZE, REFINE, para construir nuevos sustituyentes en los distintos ligandos, se buscaron orientaciones alternativas para los nuevos ligandos con el movimiento FLY, y con el movimiento TWIST se minimizaron las poses generadas por el movimiento FLY, calculando su energía de enlace y se evaluaron los ligandos generados con los movimientos WEED y SAVE (11). Este procedimiento se repitió cambiando la proporción de los movimientos para generar una mayor variedad de ligandos. Se tomaron los 10 ligandos con mayor energía de enlace de las diferentes moléculas guías, se llevaron a optimización por el método HF/3-21G*, se realizó el acoplamiento molecular entre la proteína PTP 1B y los ligandos generados, y se predijo su actividad biológica teórica.
RESULTADOS
Los resultados del acoplamiento molecular para los 40 ligandos tipo benzimidazol e imidazolina con el modelo proteico 2VEW, mostraron que todos los ligandos se posicionan en el interior de la cavidad de la proteína; además, la ubicación en el sitio de unión es muy parecida y se orientan en la misma dirección a la del ligando nativo. Sin embargo, los grupos benzimidazol e imidazolina de los ligandos estudiados se ubican en el sitio de unión de forma distinta, dependiendo de sus características fisicoquímicas y sus sustituyentes.
Las actividades biológicas, tanto experimentales como predichas, y su diferencia para ligandos tipo imidazolina y benzimidazol se representan en las tablas 1 y 2, respectivamente. Del mismo modo, las estructuras guía de los tipos de ligando, a partir de las que se derivó cada uno de los candidatos, aparecen en las figuras 2 y 3.


El modelo CoMFA fue obtenido con 40 moléculas, 32 de ellas para el conjunto de entrenamiento y 8 en el de prueba.
La mejor superposición del conjunto de moléculas estudiadas con respecto al ligando IZN15 puede apreciarse en la figura 4.

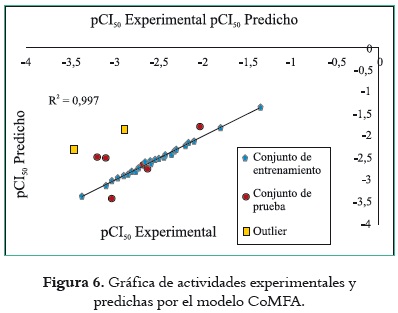
Se evaluó el poder predictivo del modelo 3DQSAR derivado del conjunto de entrenamiento mediante la predicción de la actividad biológica de las moléculas del conjunto de prueba. La contribución de los campos estéricos y electrostáticos produjo una validación cruzada de 0,659 para 10 componentes, un coeficiente de correlación no validado de 0,997, un valor de F de 748,696 y un R2 de booststraping de 0,999. La contribución de campos estéricos y electrostáticos fue 58,09% y 41,10% respectivamente, como se puede ver en la tabla 3.

Luego del acoplamiento molecular y del 3D-QSAR, se procedió a la optimización de las moléculas IZN 6, IZN9, IZN10, IZN12, IZN14, IZN 15 y BIZ 17, por tener la mayor actividad biológica de todo el grupo de moléculas, a partir de la mejor conformación obtenida en el acoplamiento molecular por medio del módulo LeapFrog de SYBYL. Del procedimiento al que fueron sometidos estos 7 ligandos se obtuvieron más de 10.000 moléculas.
La forma en que se muestran las moléculas generadas por el programa LeapFrog inicia con el nombre de la molécula precursora del lado izquierdo, seguida de un guión bajo y el número correspondiente a su posición en el momento de ser guardada por el programa. Todas las moléculas generadas mejoraron la energía de enlace con relación a su precursora (moléculas de color rojo). Además, al predecir la actividad biológica de estas moléculas se encontró que 46 de ellas (que aparecen en color azul), mejoraron su actividad biológica con respecto a su precursora, como se puede ver en la tabla 4.
DISCUSIÓN
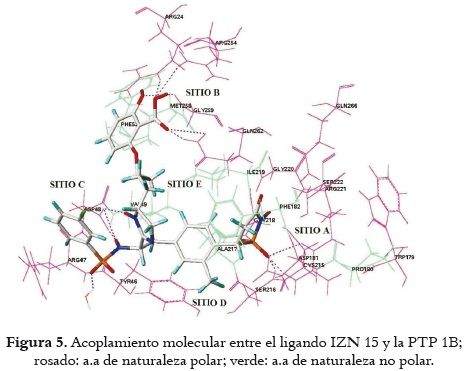
Una vez analizados los resultados del acoplamiento molecular se observó que las principales interacciones entre la PTP 1B y todos los ligandos se presentan entre el grupo isotiazolidinona y el sitio catalítico (sitio A) por medio de puentes de hidrógenos entre los aminoácidos de naturaleza polar Asp 181, Cys 215, Ser 216, Gly 220 y Arg 221 y los oxígenos presentes en este grupo. Además, se presentan uniones entre el ligando y el sitio C de la proteína por medio de los oxígenos del grupo carboxilo presentes en la cadena lateral del residuo Asp 48 (bidentado) y los nitrógenos presentes tanto en ligandos tipo imidazolina como benzimidazol. Por otro lado, interacciones estabilizantes de naturaleza no polar se dan entre los residuos Ile 219, Val 49, Ala 217 y Phe 182, y el anillo aromático adyacente a la isotiazolidinona. Todas las interacciones detalladas anteriormente son esenciales para la unión entre la enzima y los ligandos y se presentan en todas las moléculas estudiadas. La molécula IZN15 desarrolla la mayor actividad biológica del grupo de ligandos estudiados, debido a dos factores: el primero, la unión del sustituyente de la imidazolina con el sitio alostérico (sitio B); el segundo corresponde a la interacción entre el o-metilo del fenilo adyacente a la isotiazolidinona y el sitio D de la enzima, como puede observarse en la figura 5.
La figura 6 representa la recta de regresión lineal obtenida del modelo 3DQSAR a partir de actividades experimentales y predichas. En las tablas 1 y 2 se examinaron los valores atípicos (outliers), que corresponden a moléculas cuya diferencia entre actividad experimental y predicha fuera mayor de una unidad de logaritmo (log). Los ligandos INZ5 e IZN11 tuvieron valores residuales de -1,176 y -1,061, respectivamente. Una explicación para la aparición de estos outliers podría fundamentarse en la composición del conjunto de entrenamiento. Si se observa en las tablas 3 y 4, se nota que los ligandos responsables de estos valores atípicos poseen las actividades biológicas experimentales más bajas, lo cual implica que en el momento de la predicción por medio del método de regresión PLS se intente ajustar estos valores a los demás, dando lugar a grandes diferencias entre el valor predicho y el experimental de estas moléculas. Esto hace que estas moléculas sean presentadas como outliers. La razón de la pobre actividad biológica de los ligandos en cuestión puede explicarse de mejor manera por el acoplamiento molecular. En la molécula IZN 5, la configuración (R) del fenilo evita su interacción con residuos de naturaleza polar; del mismo modo, el grupo sulfona (de naturaleza polar) se encuentra presente solamente en los ligandos IZN11, IZN12 e IZN13; su presencia es favorable cuando este grupo se encuentra a dos o tres carbonos del anillo imidazolínico, dando como resultado mejores actividades biológicas; sin embargo, en la molécula IZN11, este grupo no alcanza a tener contacto con los aminoácidos de naturaleza no polar del sitio B. Ambas condiciones de estructura y configuración de estos ligandos acarrean disminución en su actividad, y por ende, un valor atípico con respecto a la predicción de su actividad.
El modelo CoMFA se representa por medio de contornos de tres dimensiones. Los contornos coloreados en el mapa representan aquellas áreas en el espacio 3D donde los cambios en los valores estéricos y electrostáticos de un compuesto se correlacionan con los cambios en su actividad biológica. Los mapas de contornos estéricos se representan en color verde (región favorable para grupos voluminosos), y amarillo (región no favorable para grupos voluminosos), mientras que los de color rojo (región favorable para sustituyentes electronegativos), y azul (región favorables para sustituyentes electropositivos), corresponden a contornos electrostáticos (10).
En los campos de contorno estérico obtenidos para las moléculas BIZ17 y BIZ19, como se representa en las figuras 7A y 7B respectivamente, la distancia entre las porciones sustituidas de estas moléculas y sus respectivos contornos varía según el tamaño del sustituyente; el poliedro amarillo de mayor tamaño, desfavorable para grupos voluminosos, se encuentra más alejado del benzimidazol en la molécula BIZ17 (CI50 = 150 nM), que posee un trifluorometilo en la posición 5, en comparación con la molécula BIZ19 (CI50 = 470 nM); la poca distancia entre el contorno de color amarillo y el sustituyente ciano, que se encuentra en posición 5 del grupo benzimidazol de dicha molécula en la figura 7B, provoca una disminución de su poder como inhibidor de la proteína PTP 1B.

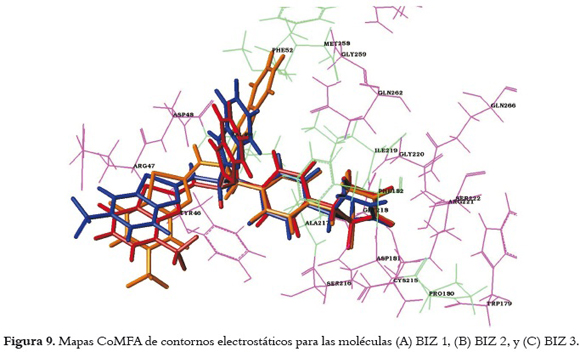
La actividad biológica también varía según la posición del sustituyente en el anillo benzotiazol, como se observa en las moléculas BIZ1, BIZ2 y BIZ3, donde fue sustituido un trifluorometilo en las posiciones 6, 5 y 4 respectivamente. A pesar de que, como se aprecia en la figura 8B (1' y 2'), la molécula BIZ2 posee contornos rojos, muy próximos al átomo de azufre y al trifluorometilo (ambos de naturaleza electronegativa) que incrementarían su actividad, también se observan contornos electrostáticos de color azul (2 y 3), que favorecen la actividad para sustituyentes electropositivos, rodeando al radical trifluorometilo, lo cual produce un descenso abrupto de las moléculas BIZ1, como se observa en la figura 8A. Por su lado, como se indica en la figura 8C, BIZ3 se ve menos afectado ya que ambas exhiben sólo una interacción desfavorable con el trifluorometilo. Los resultados obtenidos muestran contacto directo entre esta porción y el contorno azul 3 en la molécula BIZ1, y con el contorno azul 2 en la molécula BIZ3. Esta condición afecta en menor medida a la molécula BIZ1, en la que la interacción con estos contornos es menor por el distanciamiento observado entre el anillo de benzotiazol y el contorno azul 2. A pesar de que en la molécula BIZ3 el trifluorometilo (de carácter electronegativo) se encuentra en contacto directo con el contorno azul 2, también se observa que el contorno rojo 1' (favorable para átomos de alta densidad electrónica) se encuentra más próximo al anillo de benzotiazol que en la molécula BIZ2 y BIZ1, contribuyendo a la actividad biológica de la molécula. Este comportamiento también se puede explicar al observar la posición que toman los ligandos al unirse a la PTP 1B, como se aprecia en la figura 9, donde el sustituyente trifluorometilo de naturaleza polar, posicionado en el anillo de benzotiazol del ligando BIZ1 (color azul) se encuentra muy cerca de los nitrógenos pertenecientes a la cadena lateral del a.a Arg 47, aportando interacciones favorables para la estabilidad del complejo, y por ende, para la actividad biológica de la molécula. Del mismo modo se observa que el ligando BIZ3 (color rojo) se encuentra cerca del a.a Tyr 46, (no tan polar como el a.a Arg 47), ayudando a estabilizar la interacción proteína ligando; por el contrario, el ligando BIZ2 (color naranja) no tiene aminoácidos de naturaleza polar a su alrededor que puedan contribuir a la interacción del complejo, dando como resultado una disminución de la actividad biológica de la molécula.

Todas las moléculas generadas por el módulo LeapFrog del programa SYBYL mejoraron la energía de enlace con relación a su precursora, ya que la interacción entre el ligando y la proteína fue optimizada, como se puede observar en la tabla 4.
El aumento de la actividad biológica en las moléculas generadas por LeapFrog se da al adicionar sustituyentes en las moléculas precursoras en lugares donde estos grupos contribuyen a dicho aumento. Esto se ilustra en la figura 10, donde el mapa de contornos estéricos y electrostáticos generado para la molécula precursora IZN10 en el 3D QSAR presenta un contorno azul cerca de la posición 4 del anillo fluorofenilo adyacente a la sulfonamida, que indica que grupos electropositivos en esta zona generan un aumento en la actividad biológica de la molécula, lo que se comprueba al observar la molécula IZN10_224, generada en la optimización de la molécula IZN10, donde se adicionó un grupo fenilo en esta posición, como se observa en la figura 11. Además, la presencia de un contorno de color rojo (favorable para sustituyentes de naturaleza electronegativa) cerca a la sulfonamida conduce a un aumento de su capacidad como inhibidor; así mismo, en el mapa de contornos estéricos se observa que el sustituyente del grupo imidazolina se encuentra en posición opuesta al de la molécula IZN10, alejándose de los contornos amarillos (desfavorables para contornos voluminosos), lo cual conlleva aumento en la actividad biológica de la molécula por la presencia del sustituyente de la imidazolina. Estos cambios en la molécula IZN10 hacen que la molécula IZN10_224 sea tres veces más potente que su precursora.


CONCLUSIONES
Al analizar las interacciones hidropáticas entre la proteína tirosina fosfatasa 1B y los inhibidores del tipo benzoimidazol e imidazolina se puede concluir que los mapas de contorno de la molécula IZN 9 presentan más regiones favorables que los correspondientes al ligando BIZ, lo que concuerda con los valores obtenidos teóricamente en este estudio; además, fue posible explicar la actividad inhibitoria experimental de los ligandos mediante la observación de las interacciones entre el ligando y la proteína, determinando tanto los residuos responsables como las interacciones esenciales de esta unión.
A través del estudio 3D-QSAR se identifican las interacciones esenciales entre la proteína y los ligandos; además, se encuentra que el modelo generado para las moléculas acopladas con valores de q2 = 0,659 y r2 = 0,997 es estadísticamente confiable y, por tanto, tiene buen poder de correlación. Del mismo modo, los campos de contornos proporcionaron información relevante sobre la relación entre la actividad biológica de las moléculas y sus cambios estructurales.
La generación de un diseño racional de nuevos ligandos de tipo isotiazolidinona, aplicando herramientas de optimización de fármacos, nos permite proponer a los ligandos IZN6_280, IZN6_345 e IZN6_388 como buenos candidatos y sugerir su síntesis y la confirmación experimental de su actividad antidiabética, así como su evaluación farmacéutica.
AGRADECIMIENTOS
Los autores agradecen a la Universidad de Cartagena y a la Universidad del Atlántico por su permanente apoyo en la realización de esta investigación.
REFERENCIAS BIBLIOGRÁFICAS
1. Sierra L, Mendevil C. Hacia el manejo práctico de la diabetes mellitus tipo 2. 3ª ed. Bogotá, Colombia: Kimpres; 2006. 125 p. [ Links ]
2. Tuch B, Dunlop M, Proietto J. Diabetes research, a guide for postgraduates. Australia: Harwood Academic; 2000. p.1-6. [ Links ]
3. Murthy S, Kulkarni V. 3D-QSAR CoMFA and CoMSIA on protein tirosine phosphatase 1B inhibitors. Biorg Med Chem. 2002 Jul; 10 (7): 2267-2282. [ Links ]
4. Douty B, Wayland B, Ala P, Bower M, Pruitt J, Bostrom L, et al. Isothiazolidinones inhibitors of PTP 1B containing imidazoles and imidazolines. Bioorg Med Chem Lett. 2008 Jan; 18 (1): 66-71. [ Links ]
5. Sparks R, Polam P, Zhu W, Crawley M, Takvorian A, Mclaughlin E, et al. Benzothiazole benzoimidazole (S)-isothiazolidinone derivatives as protein tyrosine phosphatase-1B inhibitors. Bioorg Med Chem Lett. 2007 Feb 1; 17 (3): 736-740. [ Links ]
6. Frisch M, Trucks W, H.Schlegel, et al. GAUSSIAN 03, revisión [cd-rom]. Versión B,3, Gaussian, Inc. Pittsburg, PA USA; 2003.1 cd-rom. [ Links ]
7. The Cambridge Crystallographic Data Centre. GOLD 3.0. User's Guide [Internet]. Cambridge, UK: Cambridge Crystallographic Data Centre; 2004-2010 [updated 2004-2010; cited 2010 Apr 30]. Available from: http://www.ccdc.cam.ac.uk/support/documentation/ gold/5_0/gold/gold.1.1.html. [ Links ]
8. SYBYL Molecular Modeling System, version 7.0, Tripos Inc., St. Louis, MO, 63144-2913. [ Links ]
9. Güetto C, Ruiz JL, Torres JE, Méndez J, Vivas-Reyes R. Threedimentional quantitative structure -activity relationship studies on novel series of benzotriazine based compound acting as Src inhibitor using CoMFA and CoMSIA. Bioorg Med Chem. 2008 Mar 1; 16 (5): 2439-2447. [ Links ]
10. Nair P, Sobhia E. CoMFA based de novo design of pyridazine analogs as PTP1B inhibitors. Journal of Molecular Graphics and Modelling. 2007 Jul; 26 (1): 117-123. [ Links ]
11. SYBYL 'Ligand-Based Design Manual' version 7.3, Tripos Inc., St. Louis, MO. 63144-2913. [ Links ]
Recibido: Octubre 21 de 2009
Aceptado: Agosto 03 de 2010